Voici nos thèmes de recherche. Leur point commun réside dans l’excitation/ionisation de composés en phase gazeuse (atomes/molécules/ions) par un ou plusieurs photons. Nos travaux portent plus particulièrement sur la détection des photoproduits chargés (ions/électrons) ainsi générés, à partir desquels nous pouvons déduire le mécanisme étudié.
Ces recherches peuvent être classées en deux catégories :
– Études résolues en temps : nous utilisons une combinaison d’impulsions laser ultra-brèves (1 fs = 10⁻¹⁵ s) pour exciter le système d’intérêt et l’observer ultérieurement. En faisant varier le retard entre les deux impulsions, nous pouvons cartographier la dynamique en « temps réel », ce qui revient essentiellement à réaliser un film de la dynamique photo-induite.
– Études résolues spectralement : cette approche repose sur l’utilisation de photons d’une énergie bien définie pour accéder avec précision à des niveaux d’énergie spécifiques. La plupart de ces études sont réalisées grâce à l’utilisation du rayonnement synchrotron.
La dynamique d’une réaction chimique est l’information essentielle qui doit être obtenue pour prétendre à une vision détaillée du mécanisme de cette réaction. Le phénomène chimique étudié est photodéclenché par un laser. Cette excitation a lieu sans changement géométrique du système réactif, mais les gradients de potentiel dans l’état excité donnent au système l’impulsion nécessaire à la réaction. L’information temporelle permet d’explorer le passage du système réactif par des conformations géométriques et des configurations électroniques très éloignées de celles accessibles dans la zone de Franck-Condon.
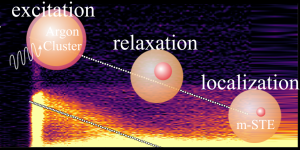
Relaxation d'excitons dans les agrégats et nanoparticules isolés
Nous avons combiné la spectroscopie de photoélectrons résolue en temps sur des espèces isolées avec le traitement de données de haut niveau pour aborder une question qui relève généralement de la science des matériaux : la dynamique de relaxation électronique vers la formation d’un exciton auto-piégé (STE, Self-trapped Exciton). Ces excitons sont des états excités courants dans les cristaux ioniques, la silice et les matrices de gaz rares. Ils sont associés à une forte déformation locale de la matrice. Les agrégats d’argon ont été pris comme système modèle. Ils sont excités initialement sur un exciton de Wannier à 14 eV et leur évolution vers la formation d’un STE a montré un type inhabituel de relaxation vibronique où l’excitation électronique de l’agrégat diminue linéairement en fonction du temps avec une vitesse de 0,59 eV/ps. La décroissance a été suivie pendant 3,0 ps, et la formation de STE s’est produite en environ 5,1 ps.
Cluster Isolated Chemical Reactions (CICR)
Etudier des réactions chimiques dans un agrégat est un moyen élégant pour étudier les effets d’un milieu réactionnel sur la dynamique d’une réaction chimique. La technique « Cluster Isolated Chemical Reaction » (CICR) est particulièrement intéressante en ce domaine, de par sa souplesse : cette technique permet de choisir un nombre connu d’atomes ou molécules réactives et de les mettre au contact d’un milieu, l’agrégat, dont la structure et la température sont connues. L’agrégat joue successivement deux rôles : en premier lieu il sert à capturer collisionnellement les réactifs, ensuite il mime le solvant. Les motivations pour isoler des réactions sur agrégat de van der Waals (ou d’hélium) sont de plusieurs types :
- Utiliser la souplesse de la technique CICR pour créer un objet d’étude difficile à obtenir autrement, et en étant sûr de renseigner spectroscopiquement sur celui-ci.
- Déterminer la stoechiométrie exacte d’une réaction.
- Etudier comment l’agrégat perturbe la réaction qui a lieu en son sein. Deux effets existent : i) mécanique en gênant des mouvements, ii) électronique en modifiant la hauteur relative des différentes surfaces de potentiel qui participent aux réactions chimiques étudiées, voire en modifiant les surfaces elles mêmes. L’interaction dipôle induit/dipôle permanent stabilise en effet les surfaces de potentiel ou les zones de surfaces de potentiel correspondant à un transfert de charge par rapport à celles pour lesquelles il n’y a pas de transfert de charge.
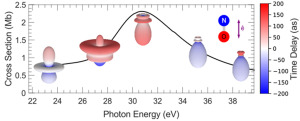
Délais de photo-ionisation attosecondes
Le délai (temps) d’émission d’un photoélectron résolu angulairement dans le référentiel moléculaire porte la signature de l’interaction de l’électron éjecté d’une orbitale moléculaire par absorption d’un photon XUV avec le potentiel électrostatique représentant la structure électronique de la molécule. La détermination de cette observable est en général l’objectif d’expériences complexes à deux photons (XUV – IR), résolues temporellement à l’échelle attoseconde. Nous avons démontré que de tels délais de photo-ionisation peuvent être déterminés par la mesure des diagrammes d’émission moléculaire des photoélectrons résolue spectralement dans le domaine XUV auprès du rayonnement synchrotron (SOLEIL). La réaction illustrant ces résultats expérimentaux et théoriques met en jeu l’interférence entre l’ionisation directe et l’ionisation résonante due à une résonance de forme, décrite par un modèle de Fano multivoies (voir figure). L’état résonnant, responsable de la modulation de la section efficace, augmente le délai d’ionisation par la rétention de l’électron avant ionisation et en modifie le diagramme d’émission.
Le délai d’ionisation peut aussi être mesuré par une approche résolue en temps, en utilisant la méthode RABBIT qui consiste à mesurer la différence de retard pris par un électron qui s’échappe d’un ion, pour différentes énergies d’ionisation. Dans le cadre d’une collaboration avec le LIDYL, l’ILM et le LCPMR, et en introduisant une approche angulaire à la mesure, nous avons pu reconstuire le film du processus d’ionisation dans l’hélium.

Interaction laser femtoseconde-nanoparticule isolée
I am text block. Click edit button to change this text. Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.
Spectroscopie des ions
L’étude des processus de photoionisation dans les espèces ioniques est indispensable pour la caractérisation et la modélisation de nombreux plasmas, qu’ils soient de laboratoire (plasmas laser, tokamak…) ou d’intérêt astrophysique (étoiles, nébuleuses…) et planétologique (ionosphères). Du point de vue fondamental, la compréhension des interactions photons-matière dans les systèmes atomiques complexes constitue un véritable défi pour les modèles théoriques qui visent à décrire les différentes interactions en compétition à l’intérieur du cortège électronique atomique. Le processus de photoionisation permet, entre autres, de suivre l’évolution de l’intensité relative des corrélations électroniques dans les séries isonucléaires en fonction de la variation de l’intensité du potentiel central.
Les expériences sur la photoionisation d’espèces ioniques ont débuté dans les années 1985 en utilisant le rayonnement synchrotron délivré par l’anneau ACO et ont été poursuivies à Super ACO, puis sur la ligne de lumière Pléiades de Soleil où le dispositif expérimental actuel MAIA dispose d’une implantation permanente. Un seul montage, situé en Allemagne, le concurrence.
Parmi les nombreux résultats ont obtenus, on peut citer l’étude du collapse de l’orbitale 4f dans l’iode, le xenon ou le Baryum en fonction du degré de charge initial de l’ion, l’étude des séries isonucléaires d’ions d’intérêt astrophysique ( C, N, O, Fe), ou l’étude des système lourds dont la modélisation nécessite l’emploi des UTA. Ces études se sont étendues à des systèmes plus complexes que sont les hydrures.
Collaborations:
E.T. Kennedy, J.P. Mosnier (Dublin College University)
C. Blancard (CEA Bruyères-le-Châtel)
B. Mc Laughlin (Harvard Smithsonian Center for Astrophysics)
T. Gorczyca (Western Michigan University)
S. Cargniato (LCPMR, Paris)

Spectromètres imageurs de vitesses (VMIS)
I am text block. Click edit button to change this text. Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.

Impulsions XUV femto et attosecondes
I am text block. Click edit button to change this text. Lorem ipsum dolor sit amet, consectetur adipiscing elit. Ut elit tellus, luctus nec ullamcorper mattis, pulvinar dapibus leo.

Goutelettes d'Hélium
MAIA
Helium nanodroplets

Rare gas clusters offer an elegant approach to capture species, such as atoms and molecules, and rapidly cool to the cluster temperature, enabling formation of weakly-bound complexes usually unattainable in gas-phase experiments. A key advantage of this method is the precise control over stoichiometry, allowing selective formation of specific complexes (b).
Among rare gas aggregates, helium droplets (a) provide a transparent “interaction-free” environment with a sub-Kelvin temperature of 0.37K. They remain transparent below 19.82 eV ensuring only embedded species interact with the light field during using valence shell excitations/ionizations.
Dopant behavior in HeDs is governed by helium affinity. Most molecular species are heliophilic and reside in the center of the droplet with low thermal energy (d1). In contrast, alkali metals and some open-shell molecules exhibit heliophobic character, residing on the droplet surface (d2). Multiple trapped species typically interact to form stable complexes in shallow potential energy wells. Excess binding energy dissipates via He atoms evaporation (b), effectively stabilizing newly formed complexes (1 He atom dissipates about ~0.6 meV). These characteristics have led some researchers to describe HeDs as “flying nano-cryo-reactors” or more modestly “ultimate spectroscopy matrices” due to their unmatched ability to host and probe reactive species with minimal perturbation.
In our laboratory, we use Helium droplet source in conjunction with ultrafast laser pulses to study a broad range of dynamics, starting from reaction dynamics between two isolated species, solvent relaxation, or gas-phase like experiments with ultracold molecules.
Time-resolved reaction dynamics
Time-Resolved Reaction Dynamics (TRRD) emerged with the development of ultrashort laser pulses and first aimed at addressing the very fundamental question: “Is it possible to follow a bimolecular reaction in real time all the way from the reactants to the products?”. The reactants and products can usually be characterized by frequency-resolved spectroscopy, but following the reaction in real time requires time-resolved methods with sufficient time resolution. TRRD focuses on tracking how chemical bonds break and form. More precisely, this field concentrates on the comprehension, the characterization and the manipulation of the forces that drive the chemical reaction. These studies are thus overcoming the static picture of bonds to project it entirely onto the dynamical aspect of the electronic and oncoming nuclear dynamics, often out of the Born-Oppenheimer approximation.
In this context, one aspect of the research focuses on the reaction dynamics between two species (atoms/molecules) embedded in a superfluid environment, hereby named Helium nanodroplet.Using a combination of ultrafast of laser pulses, we can induce and track in real time the reaction between the two species by exciting/ionizing one of them.
Relaxation dynamics
The photoexcitation of a molecule can lead to a large amount of internal energy during its relaxation through different electronic states. This relaxation typically involves numerous degrees of freedom and may lead to its fragmentation or isomerisation. In any case, this internal energy is usually a drawback as it leads to a blurring of some observables.
Consequentlty, in the case of isomerization, the different photoproducts cannot be identified properly as their photoelectron spectra overlap. This effect is well known and is usually defined as spectral congestion.
One way to circumvent this limitation, is to do the experiments in a solvent. In that case, the internal energy can be dissipated through the solvent excitation. In the case of HeDs, the low temperature of 0.37K permits to differentiate the photoproducts and also allow to preserve the molecules from fragmentation.