Développements instrumentaux
Développement d’un nouveau spectromètre pour mesures en coïncidences ultrarapides
Membres impliqués : Gildas Goldsztejn (responsable), Pierre Çarçabal.
Collaborations : Denis Cubaynes, groupe de Jérôme Palaudoux (LCPMR, Paris 6)

Le spectromètre d’électrons est de type bouteille magnétique (voir figure) dont le principe est basé sur la combinaison d’un champ magnétique fort (de l’ordre du Tesla) généré par un aimant permanent, et d’un champ homogène créé par un solénoïde le long du tube de vol qui permettent de récupérer quasiment tous les électrons générés lors de l’ionisation d’une molécule. Il est complété par un spectromètre d’ions à temps de vol dont les électrodes sont pulsées pour extraire les ions une fois que les électrons sont arrivés sur le détecteur, formant ainsi des mesures en coïncidences ions/électrons. Les molécules sont évaporées par désorption laser et refroidies par collisions avec un gaz rare issu d’une vanne pulsée. L’ensemble du dispositif permet d’étudier des molécules (fragiles ou non) en phase gazeuse ou microsolvatée à plusieurs kHz de taux de répétition.
Voir notre activité sur les mesures ultrarapides pour en savoir plus.
Développement d’une nouvelle source d’évaporation de molécules destinée aux utilisateurs du FEL à FERMI (Trieste, Italie)
Membres impliqués : Gildas Goldsztejn et Pierre Çarçabal
En collaboration avec Carlo Callegari, responsable de la ligne de lumière « Low Density Matter » du laser à électrons libres italien FERMI, nous développons actuellement une source de désorption laser, cadencée à 50 Hz pour être synchronisée avec le laser à électrons libres, accompagnée d’une expansion supersonique de gaz rare pour refroidir les molécules et ainsi les piéger dans des minima de potentiel. In fine, cette source a vocation à être ouverte aux utilisateurs de cette ligne de lumière. Ce développement est un grand pas en avant pour la communauté de la physique moléculaire car elle permettra l’ouverture vers de nombreuses molécules sous forme solide étudiées en phase gazeuse. Ce type d’évaporation douce est plus universel que l’utilisation d’un four et se prête bien notamment à l’étude de molécules fragiles qui fragmentent facilement lors de leur évaporation.
Développements théoriques
Développement d’une nouvelle méthode de simulation des spectres Auger résonants
Membres impliqués : Gildas Goldsztejn
Collaboration : Ralph Püttner (Freïe Universität, Berlin, Allemagne)
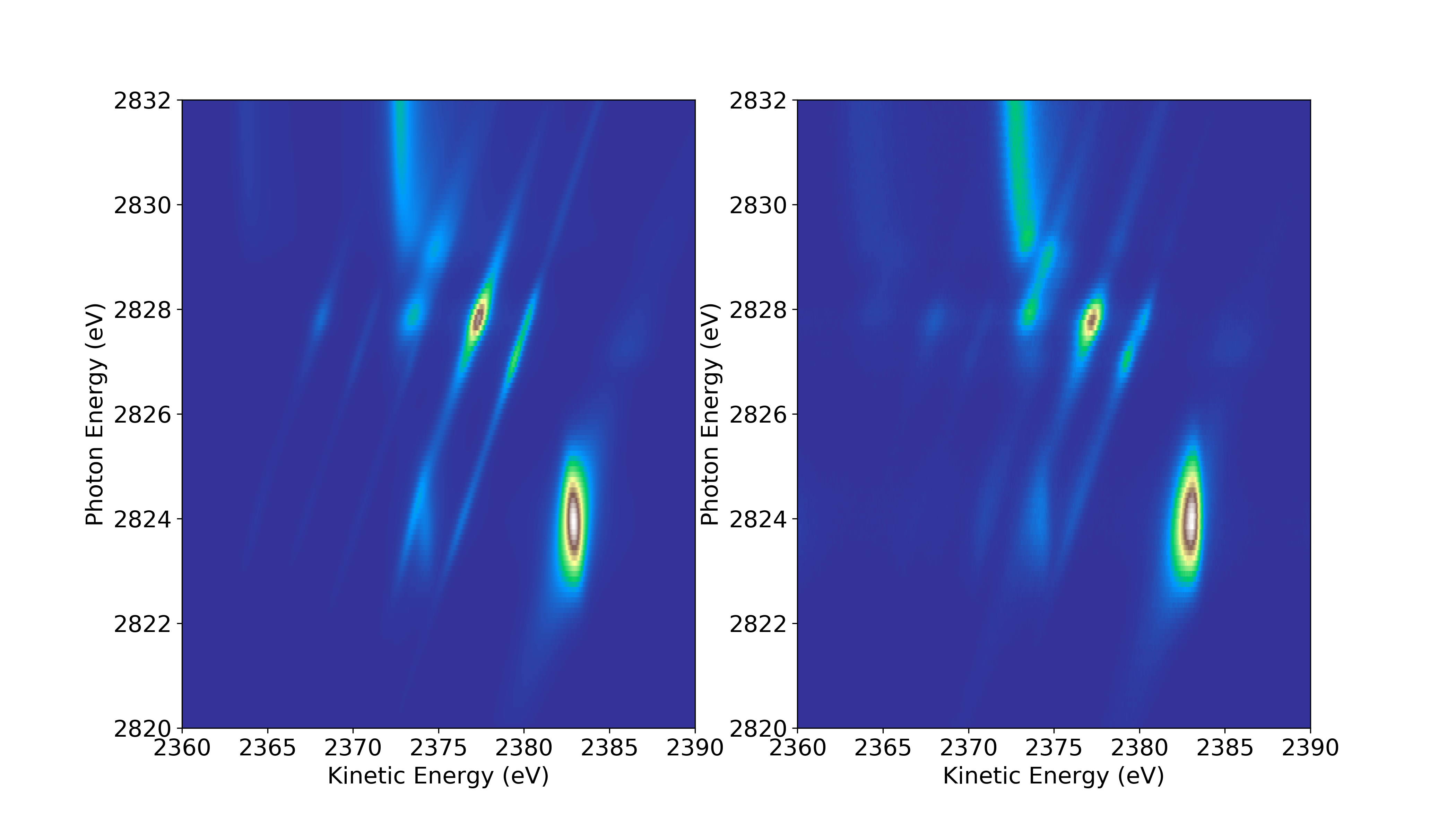
Ces dernières années nous avons développé à partir du formalisme de Kramers-Heisenberg, une méthode pour simuler les spectres Auger résonants d’abord pour des petits systèmes (diatomiques, figure de gauche) où l’on peut (presque) complètement tout calculer (https://doi.org/10.1039/D1CP05662J) et que nous avons adapté ensuite aux gros systèmes (figure de droite), où des approximations sont nécessaires, mais permettent néanmoins de reproduire qualitativement les spectres et les interpréter (https://doi.org/10.1039/D3CP01746J).
Développement d’une nouvelle méthode de simulation de la section efficace d’un état électronique relaxé
Membres impliqués : Gildas Goldsztejn
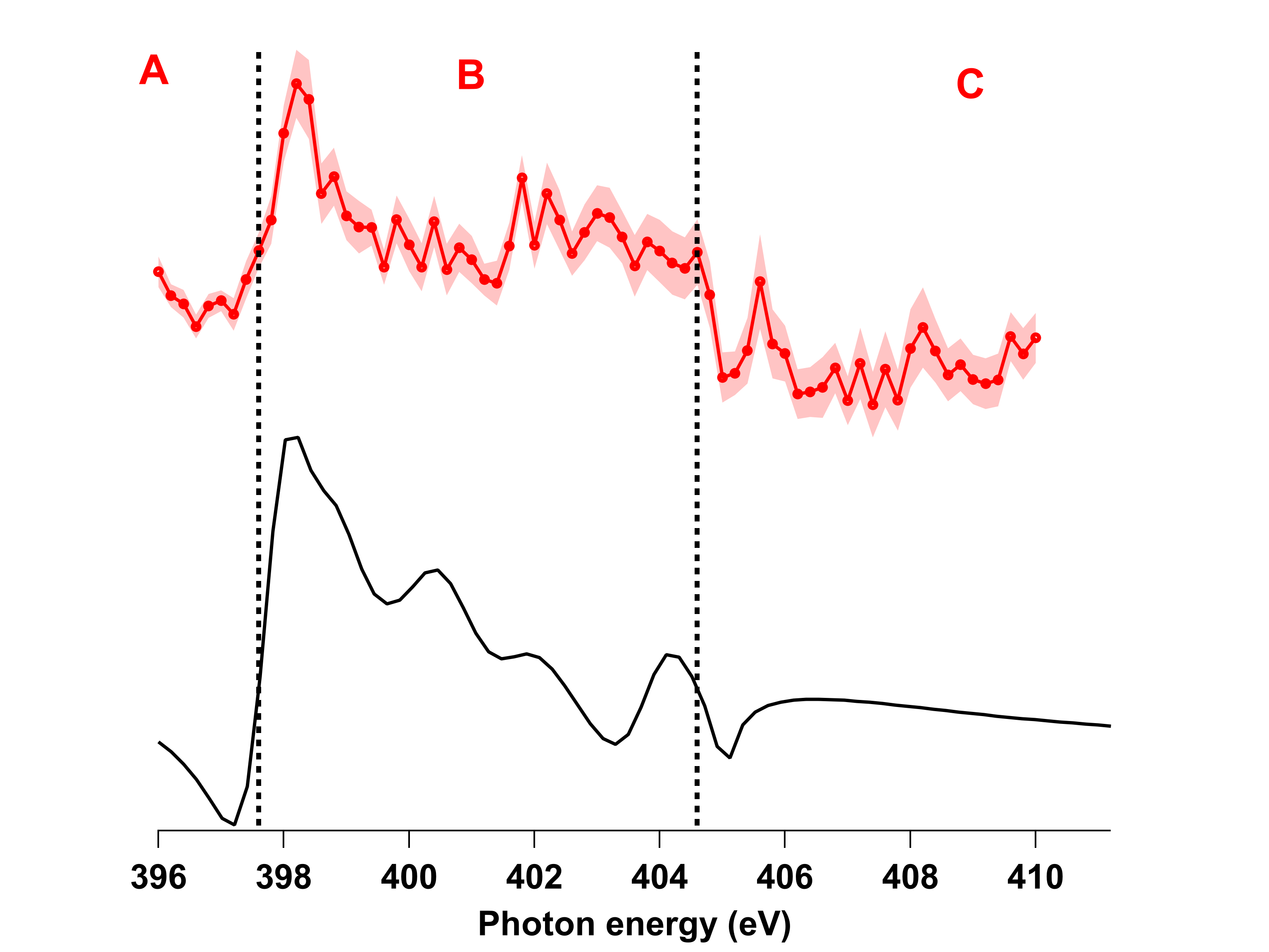
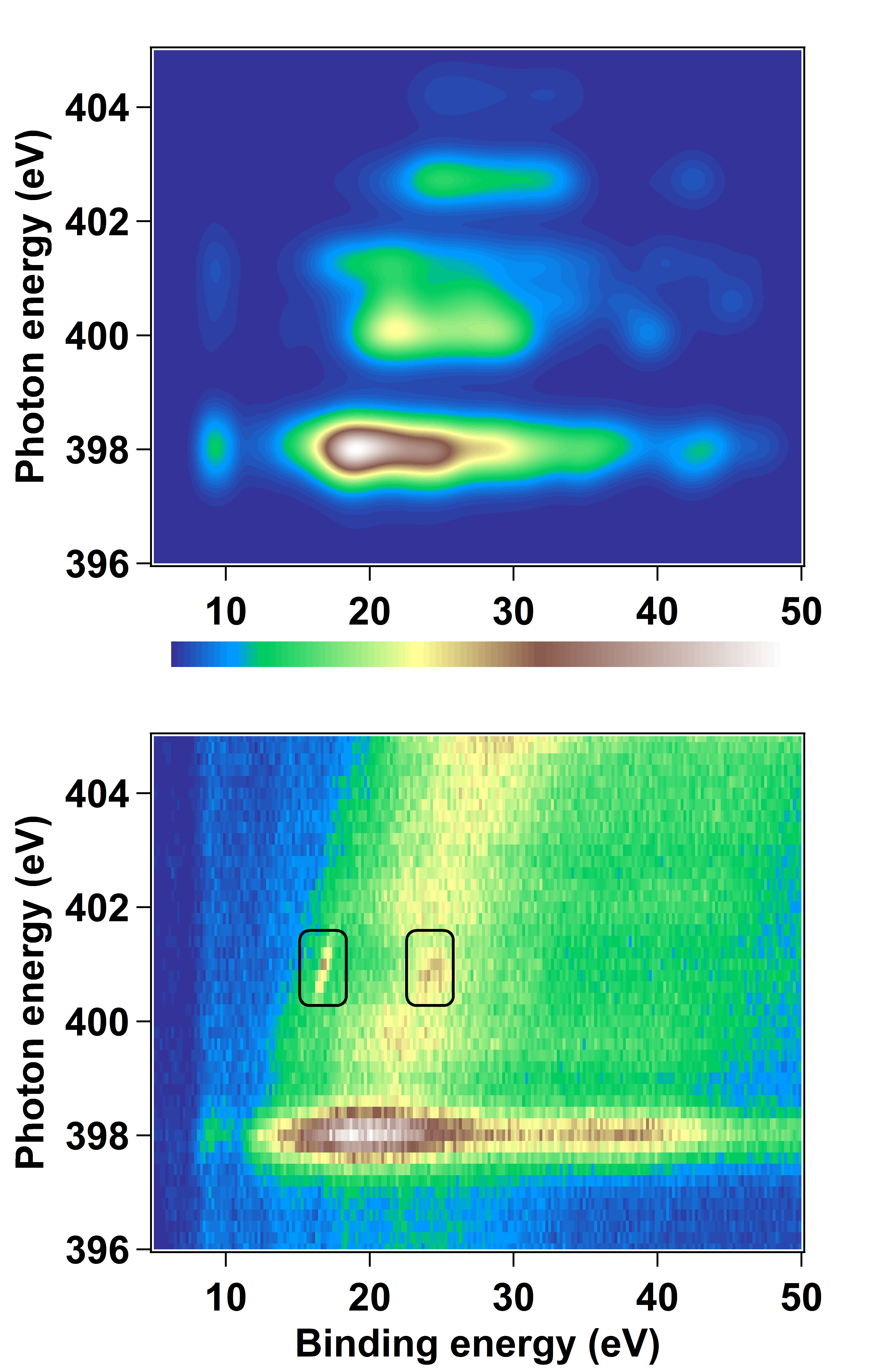
Dans la partie de notre activité de recherche sur l’étude de la relaxation d’états excités (cf. l’article ici), nous extrayons la section efficace d’un état final électronique dont nous pouvons décortiquer toutes les façons de le peupler ainsi que les possibles interférences entre différents états électroniques excités impliqués. Pour ce faire, j’ai développé une méthode pour simuler la section efficace en prenant en compte toutes les voies d’excitation menant à cet état final.
Ceci m’a permis de mettre en évidence notamment le transfert d’excitation entre la phthalocyanine et le métal, mais également l’influence des interférences de Fano entre les états discrets et continus sur la section efficace.
Calculs ab initio des spectres d’émission de photons X et de photoélectrons de molécules organométalliques
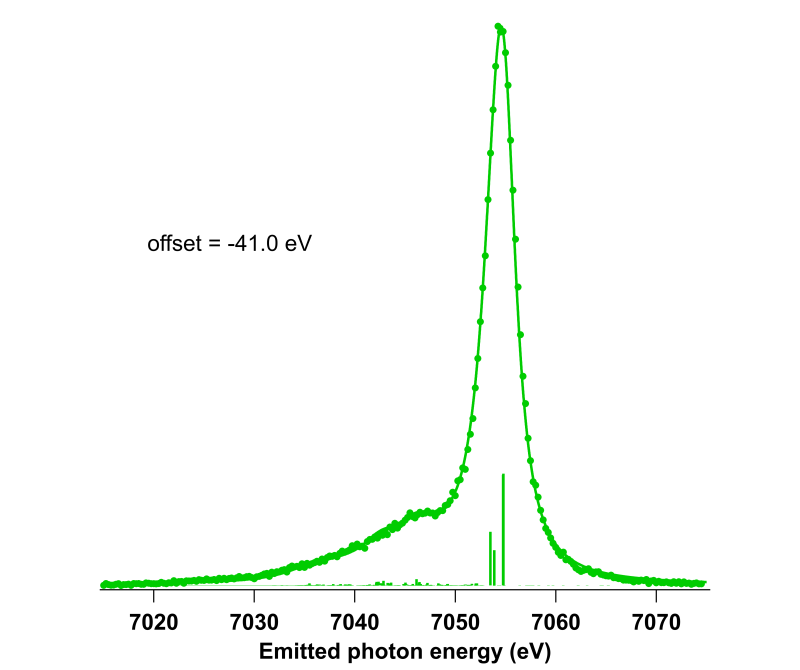 Dans le cadre d’une collaboration avec Denis Céolin (SOLEIL), Loïc Journel et Stéphane Carniato (LCPMR, Paris 6) nous avons commencé à réaliser des calculs ab-initio de l’émission X de molécules organométalliques étudiées en jet liquide.
Dans le cadre d’une collaboration avec Denis Céolin (SOLEIL), Loïc Journel et Stéphane Carniato (LCPMR, Paris 6) nous avons commencé à réaliser des calculs ab-initio de l’émission X de molécules organométalliques étudiées en jet liquide.
Particulièrement, nous nous sommes intéressés à la simulation des spectres d’émission Kβ (relaxation d’un trou en couche 1s d’un métal par un électron 3p) qui sont des spectres sensibles à l’état de spin d’une molécule, et dont la simulation n’est pas très répandue. Pour ce faire, nous avons opté pour une approche Restrictive Active Space Self Consistent Field en utilisant le logiciel de chimie quantique ORCA (figure de droite pour la molécule Fe[II](CN)6.
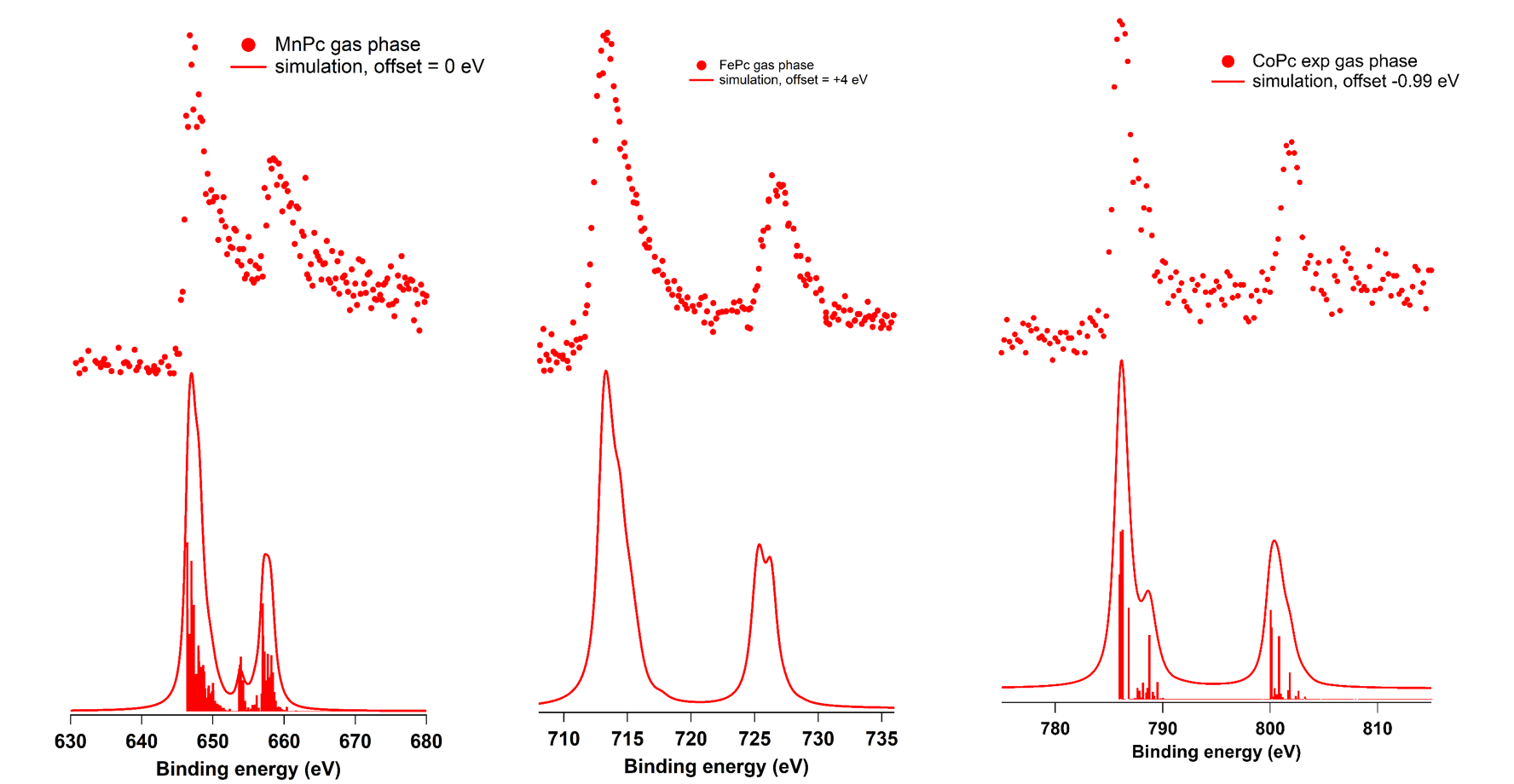
Dans le même cadre collaboratif, nous explorons une nouvelle manière de simuler les spectres d’émission de photoélectrons (XPS) de molécules organométalliques comme dans l’exemple montré sur les métallophthalocyanines, MPc (M= Mn, Fe et Co respectivement).